
Abstract
Kawasaki disease (KD) is an acute systemic vasculitis syndrome, which primarily affects in children under the age of 5 years. In 20–25% of cases, if untreated, coronary artery lesions develop, making KD the leading cause of acquired heart disease in children in both Japan and the United States. Since 1970, 19 nationwide surveys of KD in Japan have been conducted every 2 years and the data are stored in a database. Even though the etiology of KD remains unknown, despite enthusiastic research spanning more than 40 years, we have learnt a great deal about KD from this enormous database. These 19 epidemiologic studies indicate a strong genetic influence on the disease susceptibility, prompting us and other researchers to identify the responsible genes for KD by applying either the candidate gene approach or the genome-wide approach. We have employed a genome-wide linkage study using affected sibling pair data of KD in Japan and have identified several susceptibility loci. Further analysis focusing on a region of chromosome 19, where one of the linked loci was detected, identified a predisposing gene, which codes inositol 1,4,5-trisphosphate 3-kinase C (ITPKC). In this review, we summarize the cumulative knowledge regarding KD, and then outline our hypothesis of the role ITPKC plays in KD susceptibility and our trial that aims toward the implementation of personalized medicine for KD.
Similar content being viewed by others


Main
Kawasaki disease (KD; OMIM611775), alternatively called mucocutaneous lymph node syndrome (MCLS), was initially described by the Japanese pediatrician Tomisaku Kawasaki in 1967.1, 2, 3 In the early 1970s, KD was independently recognized at the University of Hawaii and the disease is now known to occur in children of all races.4
KD is basically a self-limited vasculitis syndrome affecting systemic small- and medium-sized blood vessels. The symptoms include prolonged fever unresponsive to antibiotics, nonsuppurative cervical lymphadenopathy, and changes in the skin and mucous membranes such as rash, edema, conjunctival injection, erythema of the oral cavity, lips, and palms, and desquamation of the fingertips.
Epidemiology
Started in 1970, The Japan Kawasaki Disease Research Committee has conducted nationwide surveillance of KD every 2 years. In the most recent survey for 2005–2006—the 19th survey—a total of 1543 out of 2183 hospitals responded.5 A significant number of findings were obtained as follows.
-
1
After three major outbreaks of KD in 1979, 1982, and 1986, the average annual incidence in Japan has rapidly increased to more than 10 000 patients since 2005, which is about 1.5 times higher than the number of patients of the 1979 outbreak. Owing to the decreasing birth rate in Japan, the incidence rate has been increasing more rapidly than the number of patients, reaching 188.1 (male 212.0; female 163.0) per 100 000 children aged 0–4 years in 2006, which is 2.4 times higher than that of the 1979 outbreak (Figure 1).
Figure 1 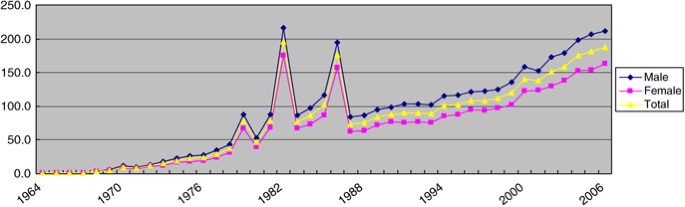
Trends in incidence rate (per 100 000 children aged 0–4 years) of Kawasaki's disease from 1964 to 2006 in Japan.5
-
2
In the 19th survey, 65.9% of the patients were under 3 years of age and peak incidence was at 6- to 8-month olds in both sexes, which is less than the peak of 9- to 11-month olds found in the 18th survey for 2003–2004.
-
3
There is a consistent predominance of males with regard to incidence. The highest male/female ratio was 1.49 in the age group of 9 to 11 month olds.
-
4
The monthly distribution showed a high peak in January and a gradual increase in summer.
-
5
Geographically, the high incidence areas were limited to certain prefectures, which varied from year to year.

KD is markedly most prevalent in Japan, followed by China, Hong Kong, Korea, and Taiwan.6, 7, 8, 9 In Hawaii, the annual incidence for Japanese Americans is similar to that for Japanese living in Japan.10 In the United States race-specific incidence rates, estimated from hospital discharge records, indicate that KD is most common among Americans of Asian and Pacific island descent (32.5/100 000 children under 5 years old), of intermediate incidence in African Americans and Hispanics (16.9 and 11.1/100 000 children under 5 years old), and lowest in Caucasians (9.1/100 000 children under 5 years old).11
Etiology and pathogenesis
The etiology of KD remains unknown. The seasonal variation, geographic distribution, and age-specific distribution strongly suggest an infectious etiology or trigger. To date, however, experiments seeking to identify the infectious agent responsible for KD, using virtually all kinds of techniques, have failed. One plausible hypothesis would be that KD is caused by some unknown, but ubiquitous infectious agent that produces the clinically apparent disease only in individuals with a genetic susceptibility. If this hypothesis is true, the frequency of the susceptible allele should be higher in Asian populations, including Japanese, than in Caucasians. Increased cytokine cascade stimulation and endothelial cell activation in acute KD are well-established findings. Taking into account the hypothesis and laboratory findings, a plausible mechanism of KD onset would be a genetically determined exaggerated immune response triggered by several different unknown microbial agents. Thus, the identification of genetic factors must surely facilitate our understanding of the disease etiology and pathophysiology.
Cardiac complications
In the 1960s in Japan, there was controversy as to whether KD was connected to subsequent cardiac complications in a significant number of cases. The first Japanese nationwide survey of KD was conducted in 1970; and from the findings of the first and second surveys, a total of 13 autopsied cases of sudden cardiac death with coronary thrombosis and aneurysm were documented,3 establishing the link between KD and coronary artery vasculitis.12, 13 It is now known that coronary artery aneurysms develop in 20–25% of untreated patients and may lead to myocardial infarction, sudden death, or ischemic heart disease.3 Until now, prevention of coronary artery lesions (CAL) is a major clinical issue to be overcome.
Treatment
The efficacy of intravenous immunoglobulin (IVIG) administered in the acute phase of KD for reducing the prevalence of CAL is well established.14, 15, 16 Patients should be treated with IVIG (2 g kg−1, single infusion) over 8–12 h, together with oral aspirin (30–50 mg kg−1 day−1). Administration of IVIG within the first 10, and preferably 7, days after the onset of fever in combination with aspirin reduces the risk of CAL to 3–5%. Approximately 15–20% of patients with KD fail to respond to initial IVIG therapy, and CAL occurs in 10–15% of patients despite this therapy. Failure to respond is typically defined as persistent fever >36 h after the completion of initial IVIG (2 g kg−1) therapy. Kobayashi et al.17 have developed a new risk score that identifies, with high sensitivity and specificity, resistance to IVIG in advance of treatment. They used seven laboratory and demographic variables available before the initiation of primary therapy.
In the case of persistent or recrudescent fever after IVIG therapy, the current treatment trial is to administer one or more repeat doses of IVIG, pulse methylprednisolone (30 mg kg−1) for 1–3 days, ulinastatin (5000 U kg−1) 3–6 times a day, together with other immunosuppressives such as cyclophosphamide, methotrexate, cyclosporin A (CsA), infliximab, or plasmapheresis. However, full evaluations of these additional therapies have not yet been completed.
Epidemiological findings indicating genetic predisposition for KD
A genetic influence on the disease susceptibility is suspected based on the following. First, as mentioned earlier, KD is over-represented among Asian and Asian-American populations.6, 7, 8, 9, 10, 11, 18 The fact that Japanese ancestries in Hawaii have higher incidence of KD than other ethnic groups and the same level of incidence as Japanese living in Japan confirms the ethnic genetic difference in susceptibility to KD. Second, KD shows familial aggregation.19, 20, 21, 22 The frequency is 10 times higher than in the general population among siblings of an index case in Japan (relative risk for siblings: λs=10). In addition, the frequency of KD was two times higher than predicted in parents of children with KD, indicating the existence of heritable factor(s).
Thus, the identification of susceptible genes could elucidate the fundamental mechanism of KD development and possibly lead to the establishment of a pharmacological therapy or preventive measure.
Candidate gene approach
One commonly used technique to identify genetic risk factors for multifactorial diseases such as KD is the candidate gene approach, which directly examines the effects of genetic variants of a possibly contributing gene by association study. To date, quite a number of genes have been examined, which are usually selected based on information regarding their function in inflammation, immune response, and other biological mechanisms. Onouchi,23 one of the authors in this article, has classified elsewhere the majority of the results of the candidate gene approach for KD and, therefore, here we briefly summarize the findings obtained from this approach.
Initial genetic studies of KD were focused on human leukocyte antigen (HLA) class I antigens. The HLA gene cluster is located at chromosome 6p21.3. To date, a number of disorders including rheumatoid arthritis, insulin-dependent diabetes mellitus, and Crohn's disease are known to be associated with the HLA subtype. There is a large ethnic difference in the distribution of HLA alleles. In Japanese KD patients, HLA-Bw22 (now referred to as Bw54) is reported to be predominant.24, 25 On the other hand, HLA-Bw51 was found to be associated with Caucasian26, 27 and Jewish KD patients,28 but no replication was obtained in Chinese29 and Korean KD patients.30 Although several other groups have studied other combinations of HLA subtypes, no consistent association has been detected so far. Considering that no significant linkage was detected near the 6p region in our genome-wide linkage analysis,31 the involvement of HLA in KD pathogenesis might be unlikely.
Among other candidate genes studied, cytokines and their receptors are in the majority. Those are chemokines and their receptors (CCL3L1,32 CCR2,34 CCR3,34 CCR5,32, 33, 34 CX3CR1,34 CXCR1,34 CXCR2,34 and MCP-135), hematopoietins (interleukin-4 (IL-4)36, 37, 38 and IL-639), IL-1 family (IL-1β,37 IL-18,40 and IL-1Ra37), IL-10 family (IL-1041), platelet-derived growth factor family (vascular endothelial growth factor (VEGF)42, 43, 44, 45 and VEGFR242), and tumor necrosis factor (TNF) family (TNF-α,46, 47, 48, 49, 50 lipoteichoic acid,47 and CD40L51, 52). Other candidates include plasma proteins (C-reactive protein53, 54, 55 and MBL253, 55, 56), matrix metalloproteinase (MMP) and their inhibitors (MMP2,58 MMP3,57, 58 MMP-9,58 MMP-12,58 MMP-13,58 and tissue inhibitors of metalloproteinase-259), enzymes related to atherosclerosis (methylenetetrahydrofolate reductase (MTHFR),60 endothelial nitric oxide synthase,61 and inducible nitric oxide synthase61), components of the renin–angiotensin system (angiotensin-converting enzyme62, 63, 64, 65 and AGTR164), and an unclassified group (CD14,66 FCGR2A,67, 68 SLC11A1,69 PLA2G7,70 UGT1A1,71 MICA,72 and HMOX171).
KD is recognized as one of the systemic inflammatory response syndromes, in which high levels of cytokines are observed.73 TNF-α is considered to play an important role in vascular injury in KD, which develops to CAL.74, 75 In accordance with this hypothesis, the usage of infliximab, the monoclonal antibody against TNF-α, has recently been found to be effective as a novel therapy for IVIG-resistant KD.76, 77, 78 Thus, genes for cytokines including TNF-α could be candidates for KD susceptibility. However, consistent results have not been obtained. This is also true for the other candidates mentioned above. Further systematic replication studies would be necessary to draw firm conclusions.
Genome-wide linkage analysis
Unlike the candidate gene approach, the genome-wide approach can be performed without any a priori assumptions about the genes. This approach therefore affords the chance to identify novel gene(s) whose function is unknown. In other words, genome-wide approaches open up the possibility of identifying multiple genetic factors that contribute to KD susceptibility with largely unknown pathogenesis. When we decided to start identifying susceptibility genes for KD about 10 years ago, the available method for the genome-wide approach was linkage analysis with affected sibling pairs. Genetic linkage is based on the phenomenon whereby alleles at loci close together on the same chromosome region will tend to be inherited together, because it is rare for a crossover to occur between the loci at meiosis. We can expect to observe co-segregation of a marker associated with a disease among affected members in the same family, and thus we can test for increased allele sharing between pairs of affected relatives, particularly affected sibling pairs. Linkage is a very long-range phenomenon; only several hundred markers may be sufficient to identify the responsible gene loci from the entire genome. Below we describe our identification of one of the susceptible genes for KD based on the results derived from genome-wide linkage analysis.
In our linkage studies we used the affected sibling pair method, a kind of non-parametric linkage study, with genome-wide distributed microsatellite markers. The total number of markers was 399 and the average interval 9.9 cM. We have recruited more than 80 families including sibling cases all over Japan. The logarithm of the odds (LOD) scores were calculated by estimating number of shared alleles identical by descent for each marker loci. The results of the genome-wide linkage analysis are shown in Figure 2. Ten chromosome regions—4q35, 5q34, 6q27, 7p15, 8q24, 12q24, 18q23, 19q13.2, Xp22, and Xq27—showed maximum LOD scores (MLS) >1.0 (Figure 2). Among them, the most significant region was 12q24 (MLS 2.69).31 These peaks, if true, implicate the location of the responsible genes for KD susceptibility. We then performed linkage disequilibrium (LD) mapping to narrow down the candidate loci and identify the genes and their functional variants.

Results of sibling pair linkage analysis. Ten chromosomal regions have positive signals of linkage (MLS>1.0). Chromosome numbers 1–22 and X are shown on the x axis.
LD mapping
LD mapping is usually performed by association study using single nucleotide polymorphisms (SNPs) near the candidate region. We selected SNPs in the vicinity of the 10–25 Mb area to the several linkage peaks from the database. A systematic case–control association study that genotyped these variants identified three significant closely located SNPs in a 150-kb LD block on the 19q13.2 region (Figure 3). A subsequent validation study using different case–control sample sets confirmed the association. We then sequenced the LD block region to find other SNPs. Identifying six additional significant SNPs through association study resulted in total nine significant SNPs. These candidate SNPs were further screened by the transmission disequilibrium test conducted on American multi-ethnic KD patient–parent trios. Of the nine significant SNPs, only four centromeric SNPs showed the same trend of association, indicating one of the four to be a functional SNP (Figure 3).79

Linkage disequilibrium mapping. Red arrowhead indicates the three SNPs initially identified. Blue arrowhead indicates SNPs identified through detailed analysis of this LD block region. Adapted with permission from Macmillan Publishers Ltd (Nat. Genet.), 79 copyright (2008).
Identification of ITPKC as a susceptibility gene for KD
As shown in Figure 3, a total of four genes—NUMBL, ADCK4, inositol 1,4,5-trisphosphate 3-kinase C (ITPKC), and FLJ41131—are located in the region of the four centromeric SNPs. Of the four genes, ITPKC was the most attractive candidate. ITPKC is a kinase of inositol 1,4,5-trsiphosphate (IP3), which is a second messenger molecule in various types of cells including T cells, macrophages, and neutrophils known to be involved in the pathogenesis of KD. IP3 is phosphorylated to become IP4 by ITPKC (Figure 4). Signals from cell surface receptors activated phospholipase C (PLC), which catalyzes the hydrolysis of plasma membrane inositol phospholipids. IP3 is one by-product of PLC-mediated phospholipid breakdown and stimulates the release of Ca2+ ions from intracellular stores and also influx of extracellular Ca2+ into the cells. In T cells, increased cytoplasmic Ca2+ binds calmodulin, and the Ca2+–calmodulin complexes activate calcineurin. Calcineurin then removes phosphates from an inactive nuclear factor of activated T cells (NFAT). Activated NFAT migrates into the nucleus, where it binds to and activates the promoters of several genes, including IL-2 (Ca2+–NFAT pathway) (Figure 5).80, 81, 82 Although a total of three isoenzymes of ITPK proteins (ITPKA, ITPKB, and ITPKC) are known to date, the importance of ITPKC in the immune system has not been identified. We investigated mRNA expression of ITPKC in a variety of tissues and revealed that ITPKC was strongly induced in stimulated peripheral blood mononuclear cells (PBMCs). Compared with the other isoenzymes, ITPKC was most abundantly expressed in PBMCs and leukemic cell lines, and most significantly induced in response to cell stimulation.79 Therefore, we speculated that ITPKC plays a significant role in the immune system and is probably related to inflammation. The finding of the predicted nuclear factor-κB binding sequence in the promoter of ITPKC in an in silico study83 is in accordance with our speculation.

ITPKC phosphorylates IP3 (left) to produce IP4 (right).

The possible role of ITPKC as a negative regulator in the Ca2+/NFAT pathway. IP3 binds to its receptor expressed on the endoplasmic reticulum (ER) membrane and causes the release of Ca2+ into the cytoplasm. Then the depletion of Ca2+ store in ER evokes a process termed as store-operated Ca2+ entry in which extracellular Ca2+ enters through calcium release-activated Ca2+ channels on the plasma membrane. Cytoplasmic Ca2+ binds calmodulin, which in turn activates calcineurin, a calmodulin-dependent phosphatase. Activated calcineurin dephosphorylates to NFAT in the cytoplasm and leads to nuclear translocation of NFAT. NFAT in the nucleus drives the transcription of genes important in T-cell activation. [Ca2+]i, intracellular free Ca2+ concentration. Panel a denotes in case of higher ITPKC activity, and panel b denotes of lower ITPKC activity.
Role of ITPKC in T-cell receptor signal transduction
We hypothesized that ITPKC negatively regulates T-cell receptor signal transduction by decreasing the amount of IP3 in the cytoplasm. If this is true, increased ITPKC expression should lead to decreased IL-2 expression, and decreased ITPKC expression should lead to increased IL-2 expression. In accordance with our hypothesis, overexpression in Jurkat cells resulted in reduced NFAT activation and IL-2 expression. On the contrary, knock-down of ITPKC by short hairpin RNA enhances NFAT activity and IL-2 expression.79 These data strongly support our hypothesis (Figures 5a and b). Through functional analyses of significant SNPs, we identified that itpkc_3 G/C, an SNP located in intron 1 of ITPKC, reduces mRNA expression of ITPKC in PBMCs down to 70% by altering splicing efficiency.79 Relatively reduced ITPKC activity associated with the C allele of itpkc_3 may enhance T-cell activation, leading to the development of KD triggered by unknown infection. Further investigation of the role of human ITPKC and biological significance of itpkc_3 in other immune cells (e.g., macrophages, B cells, and neutrophils) or non-immune cells (e.g., endothelial cells and cardiac myocytes) may elucidate a part of the pathogenesis of KD.
Prediction of CAL formation and IVIG responsiveness
As mentioned above, 15–20% of KD patients respond poorly to IVIG and these patients are at higher risk for developing CAL. The development of clinical measures to prevent CAL is thus the most important issue for pediatricians responsible for KD patients. We can assume that there are several genes related to IVIG responsiveness and CAL formation in KD, and if this is so, prediction by genotyping such genes would be beneficial in the clinical setting. We stratified our samples based on CAL formation and IVIG responsiveness. The C allele of itpkc_3 was found to be significantly more predominant in patients with CAL and those refractory to IVIG (Table 1),79 indicating that ITPKC itself is at least one such gene.
In Japan, several cases of successful CsA treatment for IVIG-resistant KD have been reported at meetings. CsA is known to bind to and inhibit the activity of calcineurin, thereby inhibiting the production of cytokines by T cells. The fact that CsA is effective indicates the importance of Ca2+/NFAT pathway activation in the pathogenesis of KD.
We are now conducting a KD prospective cohort study to determine whether or not genotyping can predict IVIG responsiveness and CAL formation, and also whether or not an association between CsA effectiveness and genotype exists. This kind of clinical trial will contribute to the establishment of evidence-based and personalized medicine in the near future.
Further research toward personalized medicine for KD
The International HapMap Consortium genotyped cell lines derived from trios of European and Yoruban descent, providing an extremely rich data set for identifying responsible genes for multifactorial diseases. On the basis of data obtained by the HapMap project completed in 2005—data that are freely available on the project's website—analysis kits for a genome-wide association study were provided by several suppliers. The fact that the genome-wide association study enables 500 000 to 1 million SNPs to be genotyped at a time has made it a mainstream technology. During the next few years, multiple susceptibility genes may be identified from such large-scale studies.
Pathways and molecular networks in which susceptibility genes identified in the genome-wide approach will provide new candidate genes. Based on our findings of the significance of the Ca2+/NFAT pathway, components of this pathway could be candidates. Through rapid progress in molecular biological technology and expanding knowledge of the human genome, we can expect the entire genetic background of KD to be clarified in the near future, bringing us closer to the availability of personalized medicine for KD.
References
Kawasaki, T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes: a clinical observation (in Japanese). Jpn. J. Allergy 16, 178–222 (1967).
Burns, J. C. Commentary: translation of Dr Tomisaku Kawasaki's original report of fifty patients in 1967. Pediatr. Infect. Dis. J. 21, 993–995 (2002).
Kawasaki, T., Kosaki, F., Okawa, S., Shigematsu, I. & Yanagawa, H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 54, 271–276 (1974).
Dean, A. G., Melish, M. E., Hicks, R. & Palumbo, N. E. An epidemic of Kawasaki syndrome in Hawaii. J. Pediatr. 100, 552–557 (1982).
Nakamura, Y., Yashiro, M., Uehara, R., Oki, I., Watanabe, M. & Yanagawa, H. Epidemiologic features of Kawasaki disease in Japan: results from the nationwide survey in 2005–2006. J. Epidemiol. 18, 167–172 (2008).
Du, Z. D., Zhao, D., Du, J., Zhang, Y. L., Lin, Y., Liu, C. et al. Epidemiological study on Kawasaki disease in Beijing from 2000 through 2004. Pediatr. Infect. Dis. J. 26, 449–451 (2007).
Huang, G. Y., Ma, X. J., Huang, M., Chen, S. B., Huang, M. R., Gui, Y. H. et al. Epidemiologic pictures of Kawasaki disease in Shanghai from 1998 through 2002. J. Epidemiol. 16, 9–14 (2006).
Chang, L. Y., Chang, I. S., Lu, C. Y., Chiang, B. L., Lee, C. Y., Chen, P. J. et al. Epidemiological features of Kawasaki disease in Taiwan, 1996–2002. Pediatrics 114, e678–e682 (2004).
Park, Y. W., Han, J. W., Park, I. S., Kim, C. H., Yun, Y. S., Cha, S. H. et al. Epidemiologic study of Kawasaki disease in Korea, 2000–2002. Pediatric. Int. 47, 382–387 (2005).
Holman, R. C., Curns, A. T., Belay, E. D., Steiner, C. A., Effler, P. V., Yorita, K. L. et al. Kawasaki syndrome in Hawaii. Pediatr. Infect. Dis. J. 24, 429–433 (2005).
Holman, R. C., Shahriari, A., Effler, P. V., Belay, E. D. & Schonberger, L. B. Kawasaki syndrome hospitalizations among children in Hawaii and Connecticut. Arch. Pediatr. Adolesc. Med. 154, 804–808 (2000).
Kato, H., Koike, S., Yamamoto, M., Ito, Y. & Yano, E. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome. J. Pediatr. 86, 892–898 (1975).
Kato, H., Sugimura, T., Akagi, T., Sato, N., Hashino, K., Maeno, Y. et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 94, 1379–1385 (1996).
Furusho, K., Kamiya, T., Nakano, H., Kiyosawa, N., Shinomiya, K., Hayashidera, T. et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet 2, 1055–1058 (1984).
Newburger, J. W., Takahashi, M., Beiser, A. S., Burns, J. C., Bastian, J., Chung, K. J. et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N. Engl. J. Med. 324, 1633–1639 (1991).
Terai, M. & Shulman, S. T. Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose. J. Pediatr. 131, 888–893 (1997).
Kobayashi, T., Inoue, Y., Takeuchi, K., Okada, Y., Tamura, K., Tomomasa, T. et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation 113, 2606–2612 (2006).
Cook, D. H., Antia, A., Attie, F., Gersony, W. M., Kamiya, T., Kato, H. et al. Results from an international survey of Kawasaki disease in 1979–82. Can. J. Cardiol. 5, 389–394 (1989).
Fujita, Y., Nakamura, Y., Sakata, K., Hara, N., Kobayashi, M., Nagai, M. et al. Kawasaki disease in families. Pediatrics 84, 666–669 (1989).
Uehara, R., Yashiro, M., Nakamura, Y. & Yanagawa, H. Kawasaki disease in parents and children. Acta Paediatr. 92, 694–697 (2003).
Uehara, R., Yashiro, M., Nakamura, Y. & Yanagawa, H. Clinical features of patients with Kawasaki disease whose parents had the same disease. Arch. Pediatr. Adolesc. Med. 158, 1166–1169 (2004).
Dergun, M., Kao, A., Hauger, S. B., Newburger, J. W. & Burns, J. C. Familial occurrence of Kawasaki syndrome in North America. Arch. Pediatr. Adolesc. Med. 159, 876–881 (2005).
Onouchi, Y. Kawasaki disease calcium/NFAT susceptibility pathway and other genetic variants linked to susceptibility and outcome. Pediatr. Res. (in press).
Matsuda, I., Hattori, S., Nagata, N., Fruse, A. & Nambu, H. HLA antigens in mucocutaneous lymph node syndrome. Am. J. Dis. Child. 131, 1417–1418 (1977).
Kato, S., Kimura, M., Tsuji, K., Kusakawa, S., Asai, T., Juji, T. et al. HLA antigens in Kawasaki disease. Pediatrics 61, 252–255 (1978).
Krensky, A. M., Berenberg, W., Shanley, K. & Yunis, E. J. HLA antigens in mucocutaneous lymph node syndrome in New England. Pediatrics 67, 741–743 (1981).
Krensky, A. M., Grady, S., Shanley, K. M., Berenberg, W. & Yunis, E. J. Epidemic and endemic HLA-B and DR associations in mucocutaneous lymph node syndrome. Hum. Immunol. 6, 75–77 (1983).
Keren, G., Danon, Y. L., Orgad, S., Kalt, R. & Gazit, E. HLA Bw51 is increased in mucocutaneous lymph node syndrome in Israeli patients. Tissue Antigens 20, 144–146 (1982).
Chang, C. C., Hawkins, B. R., Kao, H. K., Chow, C. B. & Lau, Y. L. Human leucocyte antigens in southern Chinese with Kawasaki disease. Eur. J. Pediatr. 151, 866 (1992).
Oh, J. H., Han, J. W., Lee, S. J., Lee, K. Y., Suh, B. K., Koh, D. K. et al. Polymorphisms of human leukocyte antigen genes in Korean children with Kawasaki disease. Pediatr. Cardiol. 29, 402–408 (2008).
Onouchi, Y., Tamari, M., Takahashi, A., Tsunoda, T., Yashiro, M., Nakamura, Y. et al. A genomewide linkage analysis of Kawasaki disease: evidence for linkage to chromosome 12. J. Hum. Genet. 52, 179–190 (2007).
Burns, J. C., Shimizu, C., Gonzalez, E., Kulkarni, H., Patel, S., Shike, H. et al. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J. Infect. Dis. 192, 344–349 (2005).
Jhang, W. K., Kang, M. J., Jin, H. S., Yu, J., Kim, B. J., Kim, B. S. et al. The CCR5 (-2135C/T) polymorphism may be associated with the development of Kawasaki disease in Korean children. J. Clin. Immunol. (e-pub ahead of print 15 July 2008).
Breunis, W. B., Biezeveld, M. H., Geissler, J., Kuipers, I. M., Lam, J., Ottenkamp, J. et al. Polymorphisms in chemokine receptor genes and susceptibility to Kawasaki disease. Clin. Exp. Immunol. 150, 83–90 (2007).
Jibiki, T., Terai, M., Shima, M., Ogawa, A., Hamada, H., Kanazawa, M. et al. Monocyte chemoattractant protein 1 gene regulatory region polymorphism and serum levels of monocyte chemoattractant protein 1 in Japanese patients with Kawasaki disease. Arthritis Rheum. 44, 2211–2212 (2001).
Burns, J. C., Shimizu, C., Shike, H., Newburger, J. W., Sundel, R. P., Baker, A. L. et al. Family-based association analysis implicates IL-4 in susceptibility to Kawasaki disease. Genes Immun. 6, 438–444 (2005).
Wu, S. F., Chang, J. S., Wan, L., Tsai, C. H. & Tsai, F. J. Association of IL-1Ra gene polymorphism, but no association of IL-1beta and IL-4 gene polymorphisms, with Kawasaki disease. J. Clin. Lab. Anal. 19, 99–102 (2005).
Huang, F. Y., Chang, T. Y., Chen, M. R., Lee, H. C., Chiu, N. C., Chi, H. et al. The -590 C/T and 8375 A/G interleukin-4 polymorphisms are not associated with Kawasaki disease in Taiwanese children. Hum. Immunol. 69, 52–57 (2008).
Sohn, M. H., Hur, M. W. & Kim, D. S. Interleukin 6 gene promoter polymorphism is not associated with Kawasaki disease. Genes Immun. 2, 357–362 (2001).
Hsueh, K. C., Lin, Y. J., Chang, J. S., Wan, L., Tsai, Y. H., Tsai, C. H. et al. Influence of interleukin 18 promoter polymorphisms in susceptibility to Kawasaki disease in Taiwan. J. Rheumatol. 35, 1408–1413 (2008).
Jin, H. S., Kim, H. B., Kim, B. S., Lee, J. K., Seo, E. J., Yoo, H. W. et al. The IL-10 (−627 A/C) promoter polymorphism may be associated with coronary aneurysms and low serum albumin in Korean children with Kawasaki disease. Pediatr. Res. 61, 584–587 (2007).
Kariyazono, H., Ohno, T., Khajoee, V., Ihara, K., Kusuhara, K., Kinukawa, N. et al. Association of vascular endothelial growth factor (VEGF) and VEGF receptor gene polymorphisms with coronary artery lesions of Kawasaki disease. Pediatr. Res. 56, 953–959 (2004).
Breunis, W. B., Biezeveld, M. H., Geissler, J., Ottenkamp, J., Kuipers, I. M., Lam, J. et al. Vascular endothelial growth factor gene haplotypes in Kawasaki disease. Arthritis Rheum. 54, 1588–1594 (2006).
Hsueh, K. C., Lin, Y. J., Chang, J. S., Wan, L., Tsai, Y. H., Tsai, C. H. et al. Association of vascular endothelial growth factor C-634 g polymorphism in Taiwanese children with Kawasaki disease. Pediatr. Cardiol. 29, 292–296 (2008).
Huang, F. Y., Chang, T. Y., Chen, M. R., Lee, H. C., Chi, H., Chiu, N. C. et al. Lack of association of the vascular endothelial growth factor gene polymorphisms with Kawasaki disease in Taiwanese children. J. Clin. Immunol. 28, 322–328 (2008).
Kamizono, S., Yamada, A., Higuchi, T., Kato, H. & Itoh, K. Analysis of tumor necrosis factor-alpha production and polymorphisms of the tumor necrosis factor-alpha gene in individuals with a history of Kawasaki disease. Pediatr. Int. 41, 341–345 (1999).
Quasney, M. W., Bronstein, D. E., Cantor, R. M., Zhang, Q., Stroupe, C., Shike, H. et al. Increased frequency of alleles associated with elevated tumor necrosis factor-alpha levels in children with Kawasaki disease. Pediatr. Res. 49, 686–690 (2001).
Chien, Y. H., Chang, K. W., Yang, Y. H., Lu, M. Y., Lin, Y. T. & Chiang, B. L. Association between levels of TNF-alpha and TNF-alpha promoter -308 A/A polymorphism in children with Kawasaki disease. J. Formos. Med. Assoc. 102, 147–150 (2003).
Ahn, S. Y., Jang, G. C., Shin, J. S., Shin, K. M. & Kim, D. S. Tumor necrosis factor-alpha levels and promoter polymorphism in patients with Kawasaki disease in Korea. Yonsei Med. J. 44, 1021–1026 (2003).
Cheung, Y. F., Huang, G. Y., Chen, S. B., Liu, X. Q., Xi, L., Liang, X. C. et al. Inflammatory gene polymorphisms and susceptibility to Kawasaki disease and its arterial sequelae. Pediatrics 122, e608–e614 (2008).
Onouchi, Y., Onoue, S., Tamari, M., Wakui, K., Fukushima, Y., Yashiro, M. et al. CD40 ligand gene and Kawasaki disease. Eur. J. Hum. Genet. 12, 1062–1068 (2004).
Huang, F. Y., Chang, T. Y., Chen, M. R., Chiu, N. C., Chi, H., Lee, H. C. et al. Genetic polymorphisms in the CD40 ligand gene and Kawasaki disease. J. Clin. Immunol. 28, 405–410 (2008).
Biezeveld, M. H., Kuipers, I. M., Geissler, J., Lam, J., Ottenkamp, J. J., Hack, C. E. et al. Association of mannose-binding lectin genotype with cardiovascular abnormalities in Kawasaki disease. Lancet 361, 1268–1270 (2003).
Cheung, Y. F., Huang, G. Y., Chen, S. B., Liu, X. Q., Xi, L., Liang, X. C. et al. Inflammatory gene polymorphisms and susceptibility to Kawasaki disease and its arterial sequelae. Pediatrics 122, e608–e614 (2008).
Biezeveld, M. H., Geissler, J., Weverling, G. J., Kuipers, I. M., Lam, J., Ottenkamp, J. et al. Polymorphisms in the mannose-binding lectin gene as determinants of age-defined risk of coronary artery lesions in Kawasaki disease. Arthritis Rheum. 54, 369–376 (2006).
Cheung, Y. F., Ho, M. H., Ip, W. K., Fok, S. F., Yung, T. C. & Lau, Y. L. Modulating effects of mannose binding lectin genotype on arterial stiffness in children after Kawasaki disease. Pediatr. Res. 56, 591–596 (2004).
Park, J. A., Shin, K. S. & Kim, Y. W. Polymorphism of matrix metalloproteinase-3 promoter gene as a risk factor for coronary artery lesions in Kawasaki disease. J. Korean Med. Sci. 20, 607–611 (2005).
Ikeda, K., Ihara, K., Yamaguchi, K., Muneuchi, J., Ohno, T., Mizuno, Y. et al. Genetic analysis of MMP gene polymorphisms in patients with Kawasaki disease. Pediatr. Res. 63, 182–185 (2008).
Furuno, K., Takada, H., Yamamoto, K., Ikeda, K., Ohno, T., Khajoee, V. et al. Tissue inhibitor of metalloproteinase 2 and coronary artery lesions in Kawasaki disease. J. Pediatr. 151, 155–160 (2007).
Tsukahara, H., Hiraoka, M., Saito, M., Nishida, K., Kobata, R., Tsuchida, S. et al. Methylenetetrahydrofolate reductase polymorphism in Kawasaki disease. Pediatr. Int. 42, 236–240 (2000).
Khajoee, V., Kariyazono, H., Ohno, T., Ihara, K., Mizuno, Y., Kusuhara, K. et al. Inducible and endothelial constitutive nitric oxide synthase gene polymorphisms in Kawasaki disease. Pediatr. Int. 45, 130–134 (2003).
Wu, S. F., Chang, J. S., Peng, C. T., Shi, Y. R. & Tsai, F. J. Polymorphism of angiotensin-1 converting enzyme gene and Kawasaki disease. Pediatr. Cardiol. 25, 529–533 (2004).
Shim, Y. H., Kim, H. S., Sohn, S. & Hong, Y. M. Insertion/deletion polymorphism of angiotensin converting enzyme gene in Kawasaki disease. J. Korean Med. Sci. 21, 208–211 (2006).
Fukazawa, R., Sonobe, T., Hamamoto, K., Hamaoka, K., Sakata, K., Asano, T. et al. Possible synergic effect of angiotensin-I converting enzyme gene insertion/deletion polymorphism and angiotensin-II type-1 receptor 1166A/C gene polymorphism on ischemic heart disease in patients with Kawasaki disease. Pediatr. Res. 56, 597–601 (2004).
Takeuchi, K., Yamamoto, K., Kataoka, S., Kakihara, T., Tanaka, A., Sato, S. et al. High incidence of angiotensin I converting enzyme genotype II in Kawasaki disease patients with coronary aneurysm. Eur. J. Pediatr. 156, 266–268 (1997).
Nishimura, S., Zaitsu, M., Hara, M., Yokota, G., Watanabe, M., Ueda, Y. et al. A polymorphism in the promoter of the CD14 gene (CD14/-159) is associated with the development of coronary artery lesions in patients with Kawasaki disease. J. Pediatr. 143, 357–362 (2003).
Taniuchi, S., Masuda, M., Teraguchi, M., Ikemoto, Y., Komiyama, Y., Takahashi, H. et al. Polymorphism of Fc gamma RIIa may affect the efficacy of gamma-globulin therapy in Kawasaki disease. J. Clin. Immunol. 25, 309–313 (2005).
Biezeveld, M., Geissler, J., Merkus, M., Kuipers, I. M., Ottenkamp, J. & Kuijpers, T. The involvement of Fc gamma receptor gene polymorphisms in Kawasaki disease. Clin. Exp. Immunol. 147, 106–111 (2007).
Ouchi, K., Suzuki, Y., Shirakawa, T. & Kishi, F. Polymorphism of SLC11A1 (formerly NRAMP1) gene confers susceptibility to Kawasaki disease. J. Infect. Dis. 187, 326–329 (2003).
Minami, T., Suzuki, H., Takeuchi, T., Uemura, S., Sugatani, J. & Yoshikawa, N. A polymorphism in plasma platelet-activating factor acetylhydrolase is involved in resistance to immunoglobulin treatment in Kawasaki disease. J. Pediatr. 147, 78–83 (2005).
Kanai, M., Tanabe, S., Okada, M., Suzuki, H., Niki, T., Katsuura, M. et al. Polymorphisms of heme oxygenase-1 and bilirubin UDP-glucuronosyltransferase genes are not associated with Kawasaki disease susceptibility. Tohoku J. Exp. Med. 200, 155–159 (2003).
Huang, Y., Lee, Y. J., Chen, M. R., Hsu, C. H., Lin, S. P., Sung, T. C. et al. Polymorphism of transmembrane region of MICA gene and Kawasaki disease. Exp. Clin. Immunogenet. 17, 130–137 (2000).
Furukawa, S., Matsubara, T., Jujoh, K., Yone, K., Sugawara, T., Sasai, K. et al. Peripheral blood monocyte/macrophages and serum tumor necrosis factor in Kawasaki disease. Clin. Immunol. Immunopathol. 48, 247–251 (1988).
Maury, C. P., Salo, E. & Pelkonen, P. Elevated circulating tumor necrosis factor-alpha in patients with Kawasaki disease. J. Lab. Clin. Med. 113, 651–654 (1989).
Matsubara, T., Furukawa, S. & Yabuta, K. Serum levels of tumor necrosis factor, interleukin 2 receptor, and interferon-gamma in Kawasaki disease involved coronary-artery lesions. Clin. Immunol. Immunopathol. 56, 29–36 (1990).
Weiss, J. E., Eberhard, B. A., Chowdhury, D. & Gottlieb, B. S. Infliximab as a novel therapy for refractory Kawasaki disease. J. Rheumatol. 31, 808–810 (2004).
Burns, J. C., Best, B. M., Mejias, A., Mahony, L., Fixler, D. E., Jafri, H. S. et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. J. Pediatr. 153, 833–838 (2008).
Saji, T. & Kemmotsu, Y. Infliximab for Kawasaki syndrome. J. Pediatr. 149, 426 (2006).
Onouchi, Y., Gunji, T., Burns, J. C., Shimizu, C., Newburger, J. W., Yashiro, M. et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat. Genet. 40, 35–42 (2008).
Rao, A., Luo, C. & Hogan, P. G. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747 (1997).
Crabtree, G. R. & Olson, E. N. NFAT signaling: choreographing the social lives of cells. Cell 109 (Suppl), S67–S79 (2002).
Macian, F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484 (2005).
Liu, R., McEachin, R. C. & States, D. J. Computationally identifying novel NF-kappa B-regulated immune genes in the human genome. Genome Res. 13, 654–661 (2003).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research, the Ministry of Health, Labor and Welfare Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hata, A., Onouchi, Y. Susceptibility genes for Kawasaki disease: toward implementation of personalized medicine. J Hum Genet 54, 67–73 (2009). https://doi.org/10.1038/jhg.2008.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2008.9
Keywords
This article is cited by
-
Whole-exome sequencing analysis identifies novel variants associated with Kawasaki disease susceptibility
Pediatric Rheumatology (2023)
-
Coronary artery vasculitis: assessment with cardiac multi-detector computed tomography
The International Journal of Cardiovascular Imaging (2015)
-
Single-nucleotide Polymorphism rs2290692 in the 3′UTR of ITPKC Associated With Susceptibility to Kawasaki Disease in a Han Chinese Population
Pediatric Cardiology (2012)
-
Genetic polymorphisms in Kawasaki disease
Acta Pharmacologica Sinica (2011)
-
Lack of Association between ORAI1/CRACM1 Gene Polymorphisms and Kawasaki Disease in the Taiwanese Children
Journal of Clinical Immunology (2011)
