
Abstract
Hereditary tyrosinaemia type 1 (HT-1) is a rare genetic disease caused by mutations in the gene for the enzyme fumarylacetoacetase. It usually presents with liver failure but can be manifest as chronic liver disease. Rarely, it may present with nonhepatic manifestations such as renal dysfunction, porphyria-like illness or cardiomyopathy. There is a high lifetime risk of developing hepatocellular carcinoma (HCC). Prior to the development of liver transplantation, most patients died in childhood.
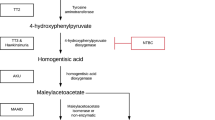
The clinical manifestations stem from the cytotoxicity of tyrosine metabolites accumulating proximal to the metabolic defect. Nitisinone acts on tyrosine metabolism upstream of the defect to prevent the production of these metabolites. Nitisinone is used in combination with a tyrosine- and phenylalanine-restricted diet.
Nitisinone has transformed the natural history of tyrosinaemia. Liver failure is controlled in 90% of patients, those with chronic liver disease improve and nonhepatic manifestations are abolished. Nitisinone is well tolerated and has few adverse effects other than a predictable rise in plasma tyrosine levels.
Nitisinone provides protection against HCC if it is started in infancy, but if commenced after the age of 2 years, a significant risk of HCC remains. Furthermore, where nitisinone is used pre-emptively, liver disease appears to be prevented, suggesting the importance of neonatal screening for tyrosinaemia where possible. Nitisinone is indicated for all children with HT-1, and liver transplantation is only indicated where nitisinone fails, or where the development of HCC is likely or suspected.

Similar content being viewed by others

References
Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol 2001; 4: 212–21
Heath SK, Gray RG, McKiernan P, et al. Mutation screening for tyrosinaemia type I. J Inherit Metab Dis 2002; 25: 523–4
Mitchell GA, Grompe M, Lambert M, et al. Hypertyrosinemia. In: Scriver CR Beaudet AR, Sly W, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001: 1777–805
van Spronsen FJ, Thomasse Y, Smit GP, et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology 1994; 20: 1187–91
Mohan N, McKiernan P, Preece MA, et al. Indications and outcome of liver transplantation in tyrosinaemia type1. Eur J Pediatr 1999; 158 Suppl. 2: S49–54
Jorquera R, Tanguay RM. Cyclin B-dependent kinase and caspase-1 activation precedes mitochondrial dysfunction in fumarylacetoacetate-induced apoptosis. FASEB J 1999; 13: 2284–98
Kubo S, Sun M, Miyahara M, et al. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc Natl Acad Sci U S A 1998; 95: 9552–7
Jorquera R, Tanguay RM. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 2001; 10: 1741–52
Luijerink MC, Jacobs SM, van Beurden EA, et al. Extensive changes in liver gene expression induced by hereditary tyrosinemia type I are not normalized by treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-l,3-cyclohexanedione (NTBC). J Hepatol 2003; 39: 901–9
Prieto-Alamo MJ, Laval F. Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I. Proc Natl Acad Sci U S A 1998; 95: 12614–8
Tschudy DP, Ebert PS, Hess RA, et al. Growth inhibitory activity of succinylacetone: studies with Walker 256 carcinosarcoma, Novikoff hepatoma and L1210 leukemia. Oncology 1983; 40: 148–54
Endo F, Kubo S, Awata H, et al. Complete rescue of lethal albino cl4CoS mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway. J Biol Chem 1997; 272: 24426–32
Al Dhalimy M, Overturf K, Finegold M, et al. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol Genet Metab 2002; 75: 38–45
Poudrier J, Lettre F, Scriver CR, et al. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab 1998; 64: 119–25
Heath SK, Gray RG, McKiernan P, et al. Mutation screening for tyrosinaemia type I. J Inherit Metab Dis 2002; 25: 523–4
Vogel A, van Den Berg I, Al-Dhalimy M, et al. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology 2004; 39: 433–43
Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis 2001; 21: 563–71
Kvittingen EA, Rootwelt H, Berger R, et al. Self-induced correction of the genetic defect in tyrosinemia type I. J Clin Invest 1994; 94: 1657–61
Demers SI, Russo P, Lettre F, et al. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol 2003; 34: 1313–20
Kim SZ, Kupke KG, Ierardi-Curto L, et al. Hepatocellular carcinoma despite long-term survival in chronic tyrosinaemia I. J Inherit Metab Dis 2000; 23: 791–804
Sokal EM, Bustos R, Van Hoof F, et al. Liver transplantation for hereditary tyrosinemia: early transplantation following the patient’s stabilization. Transplantation 1992; 54: 937–9
Lock EA, Ellis MK, Gaskin P, et al. From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-l,3-cyclohexanedione (NTBC), its toxicology and development as a drug. J Inherit Metab Dis 1998; 21: 498–506
Lindstedt S, Holme E, Lock EA, et al. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992; 340: 813–7
Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis 2000; 4: 805–14
Lock EA, Gaskin P, Ellis MK, et al. Tissue distribution of 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-l,3-dione (NTBC) and its effect on enzymes involved in tyrosine catabolism in the mouse. Toxicology 2000; 144: 179–87
Lock EA, Gaskin P, Ellis MK, et al. The effect of a low-protein diet and dietary supplementation of threonine on tyrosine and 2-(2-nitro-4-trifluoromethylbenzoyl) cyclohexane-1,3-dione-induced corneal lesions, the extent of tyrosinemia, and the activity of enzymes involved in tyrosine catabolism in the rat. Toxicol Appl Pharmacol 1998; 150: 125–32
Hall MG, Wilks MF, Provan WM, et al. Pharmacokinetics and pharmacodynamics of NTBC (2-(2-nitro-4-fluoromethylbenzoyl)-l,3-cyclohexanedione) and mesotrione, inhibitors of 4-hydroxyphenyl pyruvate dioxygenase (HPPD) following a single dose to healthy male volunteers. Br J Clin Pharmacol 2001; 52: 169–77
Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-l,3-cyclohexanedione). J Inherit Metab Dis 1998; 21: 507–17
Barkaoui E, Debray D, Habes D, et al. Favorable outcome of treatment with NTBC of acute liver insufficiency disclosing hereditary tyrosinemia type I [in French]. Arch Pediatr 1999; 6: 540–4
Croffie JM, Gupta SK, Chong SK, et al. Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics 1999; 103: 675–8
Joshi SN, Venugopalan P. Experience with NTBC therapy in hereditary tyrosinaemia type I: an alternative to liver transplantation. Ann Trop Paediatr 2004; 24: 259–65
Crone J, Moslinger D, Bodamer OA, et al. Reversibility of cirrhotic regenerative liver nodules upon NTBC treatment in a child with tyrosinaemia type I. Acta Paediatr 2003; 92: 625–8
McKiernan PJ, Baumann U, Preece MA, et al. Should we monitor lectin reactive alpha-fetoprotein in children with tyrosinaemia type 1? J Inherit Metab Dis 2005; 28 Suppl. 1: 58
Alvares F, Bussieres J-F, Dallaire L, et al. Nitisinone (NTBC) treatment of hepatorenal tyrosinaemia in Quebec. J Inherit Metab Dis 2005; 28 Suppl. 1: 49
Arora N, Stumper O, Wright J, et al. Cardiomyopathy in tyrosinaemia type 1 is common but usually resolves. J Inherit Metab Dis 2006 Feb; 29(1): 54–57
Gibbs TC, Payan J, Brett EM, et al. Peripheral neuropathy as the presenting feature of tyrosinaemia type I and effectively treated with an inhibitor of 4-hydroxyphenylpyruvate dioxygenase. J Neurol Neurosurg Psychiatry 1993; 56: 1129–32
Baumann U, Duhme V, Knerr I, et al. Lectin-reactive alpha-fetoprotein in tyrosinaemia type I [in German]. Klin Padiatr 2005; 217: 142–6
Ellaway CJ, Holme E, Standing S, et al. Outcome of tyrosinaemia type III. J Inherit Metab Dis 2001; 24: 824–32
Giardini O, Cantani A, Kennaway NG, et al. Chronic tyrosinemia associated with 4-hydroxyphenylpyruvate dioxygenase deficiency with acute intermittent ataxia and without visceral and bone involvement. Pediatr Res 1983; 17: 25–9
Gissen P, Preece MA, Willshaw HA, et al. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis 2003; 26: 13–6
Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr 1976; 88: 434–8
Perez-Cerda C, Merinero B, Sanz P, et al. Liver transplantation in nine Spanish patients with tyrosinaemia type I. J Inherit Metab Dis 1995; 18: 119–22
Dionisi-Vici C, Boglino C, Marcellini M, et al. Tyrosinaemia type 1 with early metastatic heaptocellular carcinoma: combined treatment with NTBC, chemotherapy and surgical mass removal. J Inherit Metab Dis 1997; 20 Suppl. 1: 15
van Spronsen FJ, Smit GP, Wijburg FA, et al. Tyrosinaemia type I: considerations of treatment strategy and experiences with risk assessment, diet and transplantation. J Inherit Metab Dis 1995; 18: 111–4
Pitkanen S, Salo MK, Vettenranta K, et al. Serum type III procollagen in children with type I hereditary tyrosinemia. J Pediatr Gastroenterol Nutr 1999; 29: 38–41
Hanauske-Abel HM, Popowicz A, Remotti H, et al. Tyrosinemia I, a model for human diseases mediated by 2-oxoacid-utilizing dioxygenases: hepatotoxin suppression by NTBC does not normalize hepatic collagen metabolism. J Pediatr Gastroenterol Nutr 2002; 35: 73–8
Grompe M, Lindstedt S, Al Dhalimy M, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet 1995; 10: 453–60
van Spronsen FJ, Bijleveld CM, van Maldegem BT, et al. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-l, 3-cyclohex-anedione treatment. J Pediatr Gastroenterol Nutr 2005; 40: 90–3
Acknowledgements
The author received no funding in the preparation of this article and has no conflicts of interest directly relevant to its contents.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
McKiernan, P.J. Nitisinone in the Treatment of Hereditary Tyrosinaemia Type 1. Drugs 66, 743–750 (2006). https://doi.org/10.2165/00003495-200666060-00002
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003495-200666060-00002